

“Blood is an unusual fluid”: The therapeutic antibody revolution
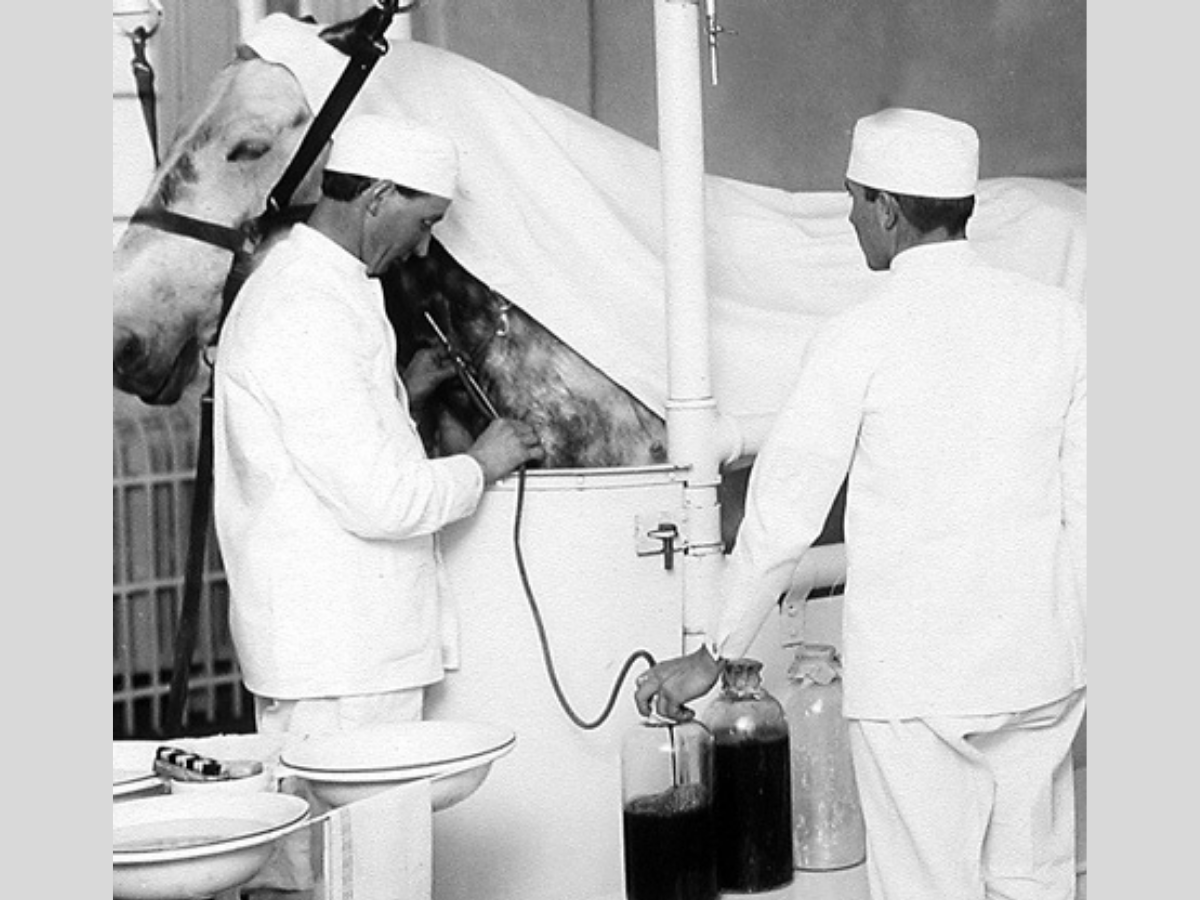
Diphtheria Antitoxin Horse: Courtesy of National Archives and Records Administration via Wikimedia Commons
Click here to listen to the full podcast episode
Replacing missing or faulty human hormones with their recombinant equivalents is a relatively straightforward way to treat disease. But by the mid-1980s, scientists were beginning to think about how they could exploit a more mysterious and unpredictable class of human proteins, antibodies.
Antibodies are small proteins produced by B cells which form part of our immune systems. They recognise and bind to foreign material like viruses, bacteria, toxins, and even rogue cells within our own bodies. Each antibody consists of four protein chains arranged in a Y shape. They have constant regions and variable regions of around 110-130 amino acids on the arms of the Y that allow the antibody to bind to different targets, or antigens.
These arms are highly specific, so each antibody only binds to one specific antigen. In theory, by varying the structure of their arms, antibodies can bind to almost any substance or antigen in the body or beyond, making them incredibly attractive for treating disease.
For example, antibodies could bind to cancer cells and flag them to the immune system or block cell growth. Antibodies can block the aberrant immune responses that lie behind autoimmune conditions like multiple sclerosis or rheumatoid arthritis. And they can also neutralise infectious agents like viruses - including the SARS-CoV-2 coronavirus that causes COVID-19.
Our understanding of antibodies and their role in immunity began back in the late 1800s when two scientists called Behring and Kitasato did experiments showing that immunity to tetanus and diphtheria toxins could be transferred between animals by extracting blood from an immunised animal, removing the cells from the blood so only a clear fluid called serum remained, then injecting this into a non-immunised animal.
Their research showed that a substance in this blood serum was able to prevent and treat infections, and it didn’t rely on the presence of cells to do so - an idea that ran against the mainstream theory at the time that cells provided immunity. They called this healing serum an “antitoxin”, though by 1891, the protective molecules within it were renamed “antibodies”.
Behring began producing antibodies from animals and using them to treat tetanus and diphtheria in humans, with successful clinical trials in 1892. A trial using antitoxin from animals to treat diphtheria in children halved the disease’s mortality rate, with the administration of the substance in the first two days of the disease almost guaranteeing recovery.
They began using horses to produce antitoxin in larger quantities, and by the early 1900s, stables around the world were set up to produce treatments for diphtheria and tetanus. Unfortunately, in 1901, 13 children from Missouri died after receiving diphtheria antitoxin from a horse that was itself sick with tetanus. This tragedy led to the regulation of biological products in the US and the founding of the FDA.
During the early 1900s, serum therapy using blood from immune animals was expanded to include treatments for measles, polio, and a range of bacterial infections. When the flu pandemic hit in 1918, serum from recovered patients was used to treat sick ones.
By 1952 scientists had isolated antibodies from human blood and began investigating how they could be used to treat disease. Soon we knew all about the structure and function of antibodies, but a huge mystery still remained: how do we generate so many different antibodies that can recognise almost any antigen?
One scientist, an Argentinian biochemist called César Milstein, who was working at the University of Cambridge, began a mission to find out.
Milstein had a theory that alterations occurring within the genetic code of the B-cells producing the antibodies were responsible for creating antibody diversity, and set about doing experiments to try and find out if he was right. His initial experiments focused on growing antibody-producing myeloma cells and looking for mutations in the genes responsible for antibody production, but this approach was laborious and not very effective.
After joining forces with Georges Köhler, a German immunologist who had moved to Cambridge to work as Milstein’s post-doc, they moved on to a different tactic. Instead of looking for gene mutations, they would grow cells that produced an antibody with very high specificity and then look for changes in the specificity of the antibody as a way of detecting new mutations.
But they had a problem: the antibody-producing cells they were using came from a type of immune cell cancer known as myeloma, but no one knew what the antibodies bound to. In fact, no one had been able to produce cell lines that made a specific antibody against a known target, or antigen. They couldn’t get a cell line off the shelf that did what they needed, so they were forced to create one.
Milstein and Köhler decided to try fusing B-cells from the spleen of a mouse that was immunised against a particular antigen with a mouse myeloma cell, hoping to combine the antibody-producing ability of the B-cell with the immortality of the myeloma cell. By 1975 Köhler had created these hybrid cells, known as hybridomas, and showed that they produced large amounts of antibodies specific to their chosen antigen - a molecule found on sheep blood cells.
Importantly, while trying to create an immortal antibody-producing cell line for their experiments, Milstein and Köhler had inadvertently created a way of making an immortal cell line capable of producing an endless supply of identical antibodies with known specificity.
So, in theory, by immunising a mouse with any antigen of your choice - from any animal or cell - you should be able to fish out immortal B-cells that churn out antibodies against that target - so-called monoclonal antibodies - which could be grown indefinitely in the lab.
When they presented their results at a meeting, one of their fellow scientists asked whether it would now be possible to make monoclonal antibodies against anything, including, as he put it, 'against my mother-in-law'.
Realising that their cells had many more potential applications than their original intended purpose, with benefits for both industry and medicine, Milstein shelved the antibody diversity problem for several years and worked on demonstrating the practical importance of monoclonal antibodies. He, Kohler, and Danish immunologist Niels Kaj Jerne shared the Nobel prize for their work on the immune system and the production of monoclonal antibodies in 1984.
Now armed with tools that allowed them to make specific antibodies, scientists began developing therapies using monoclonal antibodies. The first to be approved in 1984 was muromonab-cd3 - a mouse antibody against a protein expressed by T-cells which could prevent organ rejection in patients who had received a kidney transplant.
Unfortunately, early monoclonal antibodies lacked clinical and commercial success. Part of the problem with monoclonal antibodies created with hybridoma technologies was that the antibodies produced were mouse antibodies, which have different structures to human antibodies.
So although they were highly specific to the desired antigen, they could trigger immune responses against the antibody therapies themselves in humans, especially when used over a long period of time. Immune responses can cause rapid destruction of antibodies in the body, rendering them useless as a therapy.
To combat this problem, researchers began looking for ways to transform mouse antibodies into something that more closely resembled the human version by combining the highly specific target recognition bits of the mouse antibodies with more generic sections of human antibodies.
By the 1990s, Milstein and Köhler’s hybridoma technique had been overhauled to produce antibodies that were much more compatible with the human immune system and contained only small sections of mouse antibody structure required for specific antigen binding. The second antibody therapy to be approved did not appear until 1994. Abciximab combined sections of mouse sections and human antibodies and was designed to prevent blood clots.
Although humanisation reduced the chances that a patient’s immune system would react to the antibodies, there was still some potential for immune reactions to the antibody therapies. What’s more, antibody humanisation is complex, and the process can affect the specificity of the antibodies produced.
But Gregory Winter, a molecular biologist at the MRC in Cambridge, wanted to go further still: he wanted to create fully human antibodies. He did this using a technique called phage display, which was invented by George Smith a few years earlier and allowed him to put human antibody genes into viruses called phages, which then displayed the antibody on their surface.
Using rounds of mutagenesis and screening to allow directed evolution, Winter generated multitudes of human antibodies, which he could then screen for the best fit with his antigen, coming up with a technique that allowed him to use directed evolution to create fully human antibodies for any antigen he wanted. Smith and Winter both took shares in the 2018 Nobel prize in chemistry for their work on phage display and directed evolution of human antibodies.
The first fully human monoclonal antibody, Humira, was approved in 2002 for the treatment of rheumatoid arthritis, and quickly became a bestselling blockbuster drug.
Thanks to advances in genetic engineering and gene editing technologies like CRISPR, most new antibodies are discovered using transgenic animals, often a mouse, whose genome has been modified so that their native antibody production genes are deactivated, with human antibody genes inserted instead. As a result, when the animal is exposed to an antigen, they produce human antibodies, which can be discovered using B-cell screening, phage display or hybridoma technology.
Antibody therapy is one of the fastest-growing areas of biotechnology. To date, 570 antibody therapies have entered clinical trials, and 79 have been approved by the FDA, providing targeted therapies for diseases including various cancers, autoimmune, metabolic and infectious diseases.
Monoclonal antibodies have even been trialled against COVID-19 - most famously the monoclonal antibody cocktail made by Regeneron, which was used to treat former US President Trump. But while monoclonal antibodies do show promise for treating infectious diseases, their specificity can be their downfall, and there are concerns they don’t work against virus variants.
Even so, from a bloody start back in the 1800s to an estimated $138.6 billion dollar market by 2024, monoclonal antibody therapy is definitely one of the most promising areas of medicine, both now and in the future. Watch this space.
References:
A brief history of antibody-based therapy - Neurobiology of Disease
Development of therapeutic antibodies for the treatment of diseases - Journal of Biomedical Science
Emil von Behring: The Founder of Serum Therapy - The Nobel Prize
The Story of César Milstein and Monoclonal Antibodies - What is Biotechnology?
The Horses that Saved us From the "Strangling Angel" - Javier Yanes, OpenMind
Monoclonal Antibodies: What They Are, FDA History & Future - Nuventra


{kind=link}